Тема 11: Другий закон термодинаміки
Перший закон термодинаміки не дає відповіді на питання, в якому напрямі і до якого часу проходитиме той чи інший термодинамічний процес. Інформація про напрям термодинамічного процесу випливає із другого закону термодинаміки. Для розуміння суті цього закону ми почнемо з аналізу термодинамічних циклів.
Залежно від призначення цикли бувають прямими або зворотними. Прямими називаються цикли, які використовуються для одержання роботи завдяки теплоті. Зворотні цикли призначені для перенесення теплоти від більш холодних до більш гарячих тіл. За такими циклами працюють усі холодильні машини і теплові насоси.
Процес розширення газу в циклі здійснюють вздовж шляху, який відрізняється від шляху стиснення. Процес розширення повинен відбуватися за більш високих тисків, ніж процес стиснення.
Цикли в координатах 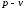 і
і зображують замкненими кривими.
зображують замкненими кривими.
ізображують замкненими кривими.
Рисунок 1.5 – Графічне зображення циклу
Процес підведення теплоти супроводжується зростанням ентропії (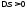 ). Щоб повернути систему в початковий стан, потрібно відвести деяку кількість теплоти
). Щоб повернути систему в початковий стан, потрібно відвести деяку кількість теплоти , внаслідок чого ентропія системи зменшується (
, внаслідок чого ентропія системи зменшується (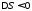 ). Отже, кількість теплоти, що перетворюється в циклі в роботу, дорівнює
). Отже, кількість теплоти, що перетворюється в циклі в роботу, дорівнює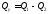 . Значення підведеної теплоти
. Значення підведеної теплоти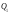 та відведеної
та відведеної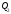 теплоти залежить від вибраного процесу. Кількість теплоти
теплоти залежить від вибраного процесу. Кількість теплоти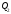 залежить від форми шляху, яким система повертається в початковий стан. Якщо система повертається за кривою 2–5–1, то
залежить від форми шляху, яким система повертається в початковий стан. Якщо система повертається за кривою 2–5–1, то 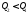 , якщо за 2–3–1, то
, якщо за 2–3–1, то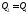 , за2–4–1,то
, за2–4–1,то 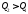 . Лише в першому випадку в циклі може бути отримана додатна робота.
. Лише в першому випадку в циклі може бути отримана додатна робота.
). Щоб повернути систему в початковий стан, потрібно відвести деяку кількість теплоти, внаслідок чого ентропія системи зменшується (). Отже, кількість теплоти, що перетворюється в циклі в роботу, дорівнює. Значення підведеної теплотита відведеноїтеплоти залежить від вибраного процесу. Кількість теплотизалежить від форми шляху, яким система повертається в початковий стан. Якщо система повертається за кривою 2–5–1, то , якщо за 2–3–1, то, за2–4–1,то . Лише в першому випадку в циклі може бути отримана додатна робота.
У зворотному циклі робота розширення відбувається за нижчих тисків і температур, ніж стиснення, і робота розширення менша, ніж робота стиснення. Такий цикл можна здійснити, лише затративши зовнішню роботу. Ефективність зворотного циклу визначається холодильним коефіцієнтом  , який чисельно дорівнює відношенню кількості теплоти, яка відбирається від холодильного джерела
, який чисельно дорівнює відношенню кількості теплоти, яка відбирається від холодильного джерела , до затраченої роботи
, до затраченої роботи
, який чисельно дорівнює відношенню кількості теплоти, яка відбирається від холодильного джерела, до затраченої роботи
Найбільш ефективним вважається такий зворотний цикл, коли за мінімальної затрати зовнішньої роботи відбирається найбільша кількість теплоти 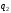 .
.
.Цикл Карно
Цей цикл був розроблений в 1824 році французьким інженером С. Карно. До складу циклу входять два ізотермічні і два адіабатні процеси, які чергуються між собою. Ці процеси оборотні, лише в цьому випадку не буде відбуватися деградація теплоти.

Рисунок 1.6 – Цикл Карно
Термічний коефіцієнт корисної дії циклу Карно, як і будь-якого теплового циклу, можна знайти за формулою
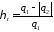
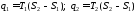
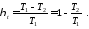
Як бачимо, значення 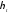 , залежить тільки від абсолютних температур тепловіддавача і теплоприймача та не залежить від властивостей робочого тіла. Це положення дістало назву теореми Карно.
, залежить тільки від абсолютних температур тепловіддавача і теплоприймача та не залежить від властивостей робочого тіла. Це положення дістало назву теореми Карно.
, залежить тільки від абсолютних температур тепловіддавача і теплоприймача та не залежить від властивостей робочого тіла. Це положення дістало назву теореми Карно.
Термічний коефіцієнт корисної дії 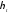 <1, тому що
<1, тому що 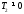 .
.
<1, тому що .
Еталоном для оцінки ефективності холодильних машин є зворотний цикл Карно, холодильний коефіцієнт якого знаходять за формулою
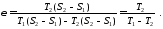
Числове значення холодильного коефіцієнта залежно від співвідношення  і
і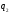 може бути як більшим, так і меншим за одиницю.
може бути як більшим, так і меншим за одиницю.
іможе бути як більшим, так і меншим за одиницю.Формулювання другого закону термодинаміки
1. Формулювання другого закону термодинаміки (Р. Клазіуса)
Теплота не може самочинно переходити від менш нагрітих тіл до більш нагрітих, або некомпенсований перехід теплоти від тіла з меншою температурою до тіла з більшою температурою неможливий.
2. Формулювання У. Томсона
Теплоту будь-якого тіла неможливо перетворити в роботу, не виконуючи ніякої іншої дії, крім охолодження цього тіла.
3. Формулювання В. Освальде
Неможливо створити вічний двигун другого роду. Під вічним двигуном другого роду розуміють двигун, який міг би працювати з одним джерелом теплоти.
Щоб вивести математичний вираз другого закону термодинаміки, використаємо вирази для ККД теплових циклів.
Термічний ККД будь-якого прямого циклу визначають за формулою

Для оборотного циклу Карно за формулою
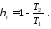
Прирівняємо ці вирази
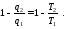
Враховуючи те, що теплота 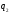 має від'ємний знак, останній вираз перепишемо
має від'ємний знак, останній вираз перепишемо
має від'ємний знак, останній вираз перепишемо
Величина 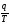 називається зведеною теплотою. Тоді отриманий результат можна сформулювати так: алгебраїчна сума зведених теплот для оборотного циклу Карно дорівнює нулю. Цей висновок може бути поширений на будь-який термодинамічний оборотний цикл.
називається зведеною теплотою. Тоді отриманий результат можна сформулювати так: алгебраїчна сума зведених теплот для оборотного циклу Карно дорівнює нулю. Цей висновок може бути поширений на будь-який термодинамічний оборотний цикл.
називається зведеною теплотою. Тоді отриманий результат можна сформулювати так: алгебраїчна сума зведених теплот для оборотного циклу Карно дорівнює нулю. Цей висновок може бути поширений на будь-який термодинамічний оборотний цикл.
Рисунок 1.7 – До виведення математичного виразу
другого закону термодинаміки
За допомогою множини адіабат розіб'ємо цей цикл на нескінченно велике число елементарних циклів, кожний з яких складається з двох ізотерм і двох адіабат і тому є елементарним циклом Карно. Для елементарного циклу маємо
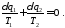
Для всього циклу можна записати

Цей вираз може бути записаним
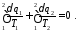
або для замкнутого контуру циклу
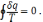
Останній вираз є математичним виразом другого закону для оборотних термодинамічних циклів.
Для необоротних циклів
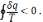
Вираз в інтегралі Клаузіуса буде дорівнювати нулю, якщо 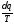 є повним диференціалом деякої функції, яка називається ентропією
є повним диференціалом деякої функції, яка називається ентропією
є повним диференціалом деякої функції, яка називається ентропією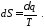
Цей вираз характеризує зміну ентропії системи, він є математичним виразом другого закону термодинаміки для оборотних термодинамічних процесів.
Розглянемо зміну ентропії в необоротних процесах. Нехай система здійснює необоротний цикл. Цей цикл складається з необоротного процесу 1-3-2 і оборотного процесу 2 - 4 -1.
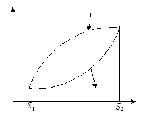
Рисунок 1.8 – Зміна ентропії в необоротних процесах
Для цього циклу інтеграл Клаузіуса має вигляд
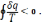
Розіб'ємо цей інтеграл на два інтеграли
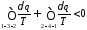
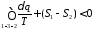
або

Продиференціювавши цей вираз, отримаємо
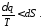
Ця нерівність є математичним виразом другого закону термодинаміки для необоротних процесів. Останнє рівняння можна записати у вигляді:

Знак рівності відноситься до оборотних термодинамічних процесів, а знак нерівності до необоротних процесів.
Натисніть на посилання.
https://www.youtube.com/watch?v=rvICc3Uuc3A
Тема 12: Термодинамічні потенціали.
Термодинамічні потенціали (термодинамічні функції) - характеристична функція в термодинаміці, спад яких в рівноважних процесах, що протікають при сталості значень відповідних незалежних параметрів, дорівнює корисної зовнішньої роботі.
Термін був введений П'єром Дюгем, Гіббс у своїх роботах використовував термін "фундаментальні функції".
Виділяють наступні термодинамічні потенціали:
- внутрішня енергія
- ентальпія
- вільна енергія Гельмгольца
- потенціал Гіббса
- великий термодинамічний потенціал
1. Визначення (для систем з постійним числом частинок)
1.1. Внутрішня енергія
Визначається відповідно до першим початком термодинаміки, як різниця між кількістю теплоти, повідомлених системі, і роботою, досконалою системою над зовнішніми тілами: .
.
1.2. Ентальпія
Визначається наступним чином: ,
,
 - тиск, а
- тиск, а  - обсяг.
- обсяг.Оскільки в Ізобаричний процесі робота дорівнює
 , Приріст ентальпії в квазістатичного изобарном процесі дорівнює кількості теплоти, отриманого системою.
, Приріст ентальпії в квазістатичного изобарном процесі дорівнює кількості теплоти, отриманого системою.1.3. Вільна енергія Гельмгольца
Також часто званий просто вільною енергією. Визначається наступним чином: ,
,
 - температура і
- температура і  - ентропія.
- ентропія.Оскільки в ізотермічному процесі кількість теплоти, отримане системою, так само
 , То спад вільної енергії в квазістатичного ізотермічному процесі дорівнює роботі, досконалою системою над зовнішніми тілами.
, То спад вільної енергії в квазістатичного ізотермічному процесі дорівнює роботі, досконалою системою над зовнішніми тілами.1.4. Потенціал Гіббса
Також званий енергією Гіббса, термодинамічним потенціалом, вільною енергією Гіббса і навіть просто вільною енергією (що може призвести до змішування потенціалу Гіббса з вільною енергією Гельмгольца): .
.
2. Термодинамічні потенціали і максимальна робота
Внутрішня енергія являє собою повну енергію системи. Однак другий початок термодинаміки забороняє перетворити всю внутрішню енергію в роботу.Можна показати, що максимальна повна робота (як над середовищем, так і над зовнішніми тілами), яка може бути отримана від системи в ізотермічному процесі, дорівнює убутку вільної енергії Гельмгольца в цьому процесі:
 ,
,
У цьому сенсі F являє собою вільну енергію, яка допускає перетворення в роботу. Частина, що залишилася внутрішньої енергії може бути названа пов'язаної.
У деяких додатках доводиться розрізняти повну і корисну роботу. Остання являє собою роботу системи над зовнішніми тілами, виключаючи середовище, в яку вона занурена. Максимальна корисна робота системи дорівнює

У цьому сенсі енергія Гіббса також є вільною.
3. Канонічне рівняння стану
Завдання термодинамічного потенціалу певної системи в певній формі еквівалентно завданню рівняння стану цієї системи.Відповідні диференціали термодинамічних потенціалів:
- для внутрішньої енергії
 ,
,
- для ентальпії
 ,
,
- для вільної енергії Гельмгольца
 ,
,
- для потенціалу Гіббса
 .
.
 ,
, ,
, ,
, .
.
 ,
,  ,
,  ,
,  - Дозволяє отримати всю інформацію про властивості системи. Так, наприклад, якщо нам задана внутрішня енергія
- Дозволяє отримати всю інформацію про властивості системи. Так, наприклад, якщо нам задана внутрішня енергія  як функція ентропії та обсягу , Що залишилися параметри можуть бути отримані диференціюванням:
як функція ентропії та обсягу , Що залишилися параметри можуть бути отримані диференціюванням:

 .
.Завдання одного з термодинамічних потенціалів як функції відповідних змінних, як записано вище, являє собою канонічне рівняння стану системи. Як і інші рівняння стану, воно справедливо лише для станів термодинамічної рівноваги. В нерівноважних станах ці залежності можуть не виконуватися.
4. Метод термодинамічних потенціалів. Співвідношення Максвелла
Метод термодинамічних потенціалів допомагає перетворювати вирази, до яких входять основні термодинамічні змінні і тим самим висловлювати такі "труднонаблюдаемие" величини, як кількість теплоти, ентропію, внутрішню енергію через вимірювані величини - температуру, тиск і обсяг та їх похідні.Розглянемо знову вираз для повного диференціала внутрішньої енергії:
 .
.
 .
.
 і
і  , Тому
, Тому .
.
 ,
, ,
, .
.
5. Системи зі змінним числом частинок. Великий термодинамічний потенціал
Хімічний потенціал ( μ ) Компонента визначається як енергія, яку необхідно затратити для того, щоб додати в систему нескінченно мале молярне кількість цього компонента. Тоді вирази для диференціалів термодинамічних потенціалів можуть бути записані так: ,
, ,
, ,
, .
.
 ,
, ,
, ,
, .
.
 , З останнього виразу випливає, що
, З останнього виразу випливає, що ,
,
Для великого канонічного ансамблю (тобто для статистичного ансамблю станів системи з змінним числом частинок і рівноважним хімічним потенціалом) може бути визначений великий термодинамічний потенціал, що зв'язує вільну енергію з хімічним потенціалом:
 ;
;
6. Потенціали і термодинамічна рівновага
У стані рівноваги залежність термодинамічних потенціалів від відповідних змінних визначається канонічним рівнянням стану цієї системи. Однак у станах, відмінних від рівноважного, ці співвідношення втрачають силу. Тим не менш, для нерівноважних станів термодинамічні потенціали також існують.Таким чином, при фіксованих значеннях своїх змінних потенціал може приймати різні значення, одне з яких відповідає стану термодинамічної рівноваги.
Можна показати, що в стані термодинамічної рівноваги відповідне значення потенціалу мінімально. Тому рівновага є стійким.
Наведена нижче таблиця показує, мінімуму будь потенціалу відповідає стан стійкої рівноваги системи із заданими фіксованими параметрами.
| фіксовані параметри | термодинамічний потенціал |
|---|---|
Комментарии
Отправить комментарий